

.
Stephen Senn
Head of Competence Center
for Methodology and Statistics (CCMS)
Luxembourg Institute of Health
Twitter @stephensenn
Being a statistician means never having to say you are certain
A recent discussion of randomised controlled trials[1] by Angus Deaton and Nancy Cartwright (D&C) contains much interesting analysis but also, in my opinion, does not escape rehashing some of the invalid criticisms of randomisation with which the literatures seems to be littered. The paper has two major sections. The latter, which deals with generalisation of results, or what is sometime called external validity, I like much more than the former which deals with internal validity. It is the former I propose to discuss.
The trouble starts, in my opinion, with the discussion of balance. Perfect balance is not, contrary to what is often claimed a necessary requirement for causal inference, nor is it something that randomisation attempts to provide. Conventional analyses of randomised experiments make an allowance for imbalance and that allowance is inappropriate if all covariates are balanced. If you analyse a matched-pairs design as if it were completely randomised, you fail that question in Stat 1. (At least if I am marking the exam.) The larger standard error for the completely randomised design is an allowance for the probable imbalance that such a design will have compared to a matched-pairs design.
This brings me on to another criticism. D&C discuss matching as if it were somehow an alternative to randomisation. But Fisher’s motto for designs can be expressed as, “block what you can and randomise what you can’t”. We regularly run cross-over trials, for example, in which there is blocking by patient, since every patient receives each treatment, and also blocking by period, since each treatment appears equally often in each period but we still randomise patients to sequences.
Part of their discussion recognizes this but elsewhere they simply confuse the issue, for example discussing randomisation as if it were an alternative to control. Control makes randomisation possible. Without control, there is no randomisation. Randomisation makes blinding possible, without randomisation there can be no convincing blinding. Thus in order of importance they are, control, randomisation and blinding but to set randomisation up as some alternative to control is simply misleading and unhelpful.
Elsewhere they claim, “the RCT strategy is only successful if we are happy with estimates that are arbitrarily far from the truth, just so long as errors cancel out over a series of imaginary experiments” but this is not what RCTs rely on. The mistake is in becoming fixated with the point estimate. This will, indeed be in error but any decent experiment and analysis will deliver an estimate of that error, as, indeed, they concede elsewhere. Being a statistician is never having to say you are certain. To prove a statistician is a liar you have to prove that the probability statement is wrong. That is harder than it may seem.
They correctly identify that when it comes to hidden covariates it is the totality of their effect that matters. In this, their discussion is far superior to the indefinitely many confounders argument that has been incorrectly proposed by others as being some fatal flaw. (See my previous blog Indefinite Irrelevance). However, they then undermine this by adding “but consider the human genome base pairs. Out of all those billions, only one might be important, and if that one is unbalanced, the result of a single trial can be ‘randomly confounded’ and far from the truth”. To which I answer “so what?”. To see the fallacy in this argument, which simultaneously postulates a rare event and conditions on its having happened, even though it is unobserved, consider the following. I maintain that if a fair die is rolled six times, the probability of six sixes in a row will be 1/46,656 and so rather rare. “Nonsense” say D&C, “suppose that the first five rolls have each produced a six, it will then happen one in six times and so is really not rare at all”.
I also consider that their simulation is irrelevant. They ask us to believe that if 100 samples of size 50 are taken from a log-Normal distribution and then for each sample, the values are permuted 1000 times to 25 in the control and 25 in the experimental group the type I error rate for a nominal 5% using the two-sample t-test will be 13.5%. In view of what is known about the robustness of the t-test under the null hypothesis (there is a long literature going back to Egon Pearson in the 1930s), this is extremely surprising and as soon as I saw it I disbelieved it. I simulated this myself using 2000 permutations, just for good measure, and found the distribution of type one error rates in the accompanying figure.
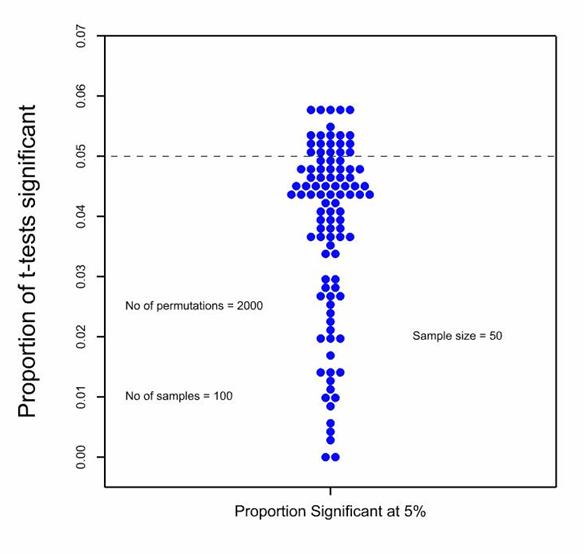 Each dot represents the type I error rate over 2000 permutations for one of the 100 samples. It can be seen that for most of the samples the proportion of significant t-tests is less than the nominal 5% and in fact, the average for the simulation is 4%. It is, of course, somewhat regrettable that some of the values are above 5% and, indeed, five of them have got a value of nearly 6% but if this worries you, the cure is at hand. Use a permutation t-test rather than a parametric one. (For a history of this approach, see the excellent book by Mielke et al [2].) Don’t confuse the technical details of analysis with the randomisation. Whatever you do for analysis, you will be better off for having randomised whatever you haven’t blocked.
Each dot represents the type I error rate over 2000 permutations for one of the 100 samples. It can be seen that for most of the samples the proportion of significant t-tests is less than the nominal 5% and in fact, the average for the simulation is 4%. It is, of course, somewhat regrettable that some of the values are above 5% and, indeed, five of them have got a value of nearly 6% but if this worries you, the cure is at hand. Use a permutation t-test rather than a parametric one. (For a history of this approach, see the excellent book by Mielke et al [2].) Don’t confuse the technical details of analysis with the randomisation. Whatever you do for analysis, you will be better off for having randomised whatever you haven’t blocked.
Why does my result differ from theirs? It is hard for me to work out exactly what they have done but I suspect that it is because they have assumed an impossible situation. They are allowing that the average treatment effect for the millions of patients that might have been included is zero but then sampling varying effects (that is to say the difference the treatment makes), rather than merely values (that is to say the reading for given patients), from this distribution. For any given sample the mean of the effects will not be zero and so the null-hypothesis will, as they point out, not be true for the sample, only for the population. But in analysing clinical trials we don’t consider this population. We have precise control of the allocation algorithm (who gets what if they are in the trial) and virtually none over the presenting process (who gets into the trial) and the null hypothesis that we test is that the effect is zero in the sample not in some fictional population. It may be that I have misunderstood what they are doing but I think that this is the origin of the difference.
This is an example of the sort of approach that led to Neyman’s famous dispute with Fisher. One can argue about the appropriateness of the Fisherian null hypothesis, “the treatments are the same”, but Neyman’s “the treatments are not the same but on average they are the same” is simply incredible[3]. As D&C’s simulation shows, as soon as you allow this, you will never find a sample for which it is true. If there is no sample for which it is true, what exactly are the remarkable properties of the population for which it is true? D&C refer to magical thinking about RCTs dismissively but this is straight out of some wizard’s wonderland.
My view is that randomisation should not be used as an excuse for ignoring what is known and observed but that it does deal validly with hidden confounders[4]. It does not do this by delivering answers that are guaranteed to be correct; nothing can deliver that. It delivers answers about which valid probability statements can be made and, in an imperfect world, this has to be good enough. Another way I sometimes put it is like this: show me how you will analyse something and I will tell you what allocations are exchangeable. If you refuse to choose one at random I will say, “why? Do you have some magical thinking you’d like to share?”
Acknowledgement
My research on inference for small populations is carried out in the framework of the IDeAl project http://www.ideal.rwth-aachen.de/ and supported by the European Union’s Seventh Framework Programme for research, technological development and demonstration under Grant Agreement no 602552.
References
- Deaton A, Cartwright N. Understanding and misunderstanding randomized controlled trials. Social Science and Medicine 2017.
- Berry KJ, Johnston JE, Mielke PWJ. A Chronicle of Permutation Statistical Methods. Springer International Publishing Limited Switzerland: Cham, 2014.
- Senn SJ. Added Values: Controversies concerning randomization and additivity in clinical trials. Statistics in Medicine 2004; 23: 3729-3753.
- Senn SJ. Seven myths of randomisation in clinical trials. Statistics in Medicine 2013; 32: 1439-1450.


")


Stephen: Thanks so much for the guest post. I’ve been in RCT discussions with them, but I’m still taken aback that they could write such an extreme claim as “the RCT strategy is only successful if we are happy with estimates that are arbitrarily far from the truth, just so long as errors cancel out over a series of imaginary experiments”. Do they really say that? I’ll have to read and study their article
Beautifully written Stephen and just a dynamite message to boot.
Stephen:
I was asked to comment on the Deaton and Cartwright paper and here is what I wrote: http://www.stat.columbia.edu/~gelman/research/published/causal_ssm.pdf
Here are some excerpts:
‘I agree with Deaton and Cartwright that randomized trials are often overrated. . . .
One way to get a sense of the limitations of controlled trials is to consider the conditions under which they can yield meaningful, repeatable inferences. . . . It is difficult to expect these conditions to be satisfied without good substantive understanding. As Deaton and Cartwright put it, “when little prior knowledge is available, no method is likely to yield well-supported conclusions.” Much of the literature in statistics, econometrics, and epidemiology on causal identification misses this point, by focusing on the procedures of scientific investigation—in particular, tools such as randomization and p-values which are intended to enforce rigor—without recognizing that rigor is empty without something to be rigorous about. . . .
This is not to say that existing investigations in social science, policy, and medicine are atheoret- ical or lacking in scientific content. Rather, there is a disconnect between the design of treatments (based on theory and the qualitative integration of the literature) and their evaluation . . . As Deaton and Cartwright note, the traditional focus in statistics and econometrics on average treatment effects (and the corresponding focus in epidemiology on single-number summaries such as hazard ratios) is misleading . . .
Randomized controlled trials have problems, but the problem is not with the randomization and the control—which do give us causal identification, albeit subject to sampling variation and relative to a particular local treatment effect. So really we’re saying at all empirical trials have problems . . . Once we recognize the importance of diverse sources of data, statistics can be helpful in making decisions and quantifying uncertainty. . . .’
—
In my discussion of Deaton and Cartwright’s paper, I completely ignored the internal-validity part—the stuff that you talk about above—because it was all expressed in terms of average treatment effects, and for reasons of external validity, I don’t think the average treatment effects, as usually defined in the causal inference literature, are so useful.
So I guess I think you (Stephen) are focusing on the less interesting and less important part of that paper. It may well be that Deaton and Cartwright made mistakes in that section, but I would not like that to detract from what I consider their larger, more important points.
Andrew: Thanks for your comment. I haven’t had the chance to read their paper (nor the comments to which you link), but I’ve been in several conferences and workshops with one (Cartwright) or both of the authors where the focus was on randomization, usually in development economics. I think it’s important to evaluate their arguments in relation to the alternative methods they endorse-based on statistical modeling. In many fields, it’s hard to see why you wouldn’t do both, if you could. The reason so-called internal validity matters is that randomization isn’t intended to supply external validity. Of course, the common experiments we see in areas like psychology presume that the randomized allocation of treatments, which is the basis for the statistical analysis, also renders the results relevant for generalizing. But that’s a mistake. I’ll say more after I’ve read the relevant items.
Deborah:
It is my impression that one of the big problems in quantitative social science and biomedical research is the attitude that, if a study has “identification” (ideally from randomization), it is rigorous. As I wrote in my above-linked response to Deaton and Cartwright, I think all their criticisms of randomized studies also apply to non-randomized studies. The difference is that, with non-randomized studies (or, in the case of economics, studies without some conventional method of identification, whether it be randomization or instrumental variables or difference in differences or regression discontinuity or matching or whatever), researchers are more appropriately cautious in their interpretation of findings. Once randomization (or other identification strategies) go in, researchers often seem to turn off their brains. And I agree with Deaton and Cartwright that this is not appropriate, that just cos a study is randomized it doesn’t mean you can start drawing all sorts of external conclusions from it. The point is obvious but needs to be made, if you look at so much of current research practice.
Andrew: I agree with your above comment and your commentary: “Where does this all leave us? “Randomized controlled trials have problems, but the problem is
not with the randomization and the control—which do give us causal identification, albeit subject
to sampling variation and relative to a particular local treatment effect. So really we’re saying at all
empirical trials have problems, a point which has arisen many times in discussions of experiments
and causal reasoning in political science”.
The supposition that because you’ve randomized “treatments,” enabling a statistical significance assessment, that the results are relevant for a research claim is wrong–let alone may one extrapolate to other populations. Granted too, in some cases, as you say, that once “randomization (or other identification strategies) go in, researchers often seem to turn off their brains”–or at least think they’re protected from fallacies. Reading into the significant or non-significant result with one or another presupposed theory is problematic, which is why Fisher demanded more for genuine effects (replication), and more still for causal inference (making your theories elaborate and varied).
However, I think it would be highly problematic to argue that since randomized studies are also open to fallacies that we might as well do non-randomized studies. I’m not saying they argue this (I will study their paper when I return from travels). Even Savage (whom they mention as denying the necessity for randomization) and other subjective Bayesians (Kadane) have worked very hard to find ways to justify it, despite the conflict with the Likelihood Principle.
In today’s world where statistical inference methods are often blamed for non-replication, rather than violations of experimental design,data-dependent selection effects, and well-known fallacies of statistical inference, and the “21 century cures” act* says, or seems to say, we do not need randomization, it’s very important to be clear on just what’s being blamed, and showing why alternative methods will do better.
*https://rejectedpostsofdmayo.com/2017/11/08/you-are-no-longer-bound-to-traditional-clinical-trials-21st-century-cures/
Andrew: I agree with you that the second part is important and should not be overlooked. Nevertheless, I think that the first past was confused and that it was important to make the counterargument because, for reasons I hope to make clear below, although I believe that generalisability is a serious issue affecting studies,, I think that it is a less serious issue (at least where clinical trials are concerned) than is ‘internal’ validity.
First, I think it is worth noting, that the issue of external validity has long been discussed by statisticians. As long ago as 1938, Yates and Cochran (1) drew attention to the fact that ‘scientific’ research was easier than ‘technical’ research. By the former they meant causal investigations relying on control and randomisation and by the latter they meant attempting to establish practical implications for implementing such findings.
Second, however, I do not consider that the discussion by Deaton and Cartwright in their first part is correct. The billion base claim, for example, cannot be allowed to stand. They are postulating something they could not possibly hope to simulate. I often put it like this. Suppose a model as complicated as you like based on some exquisite detailed knowledge of which you claim to be in possession. Can this knowledge amount to anything more than being able to predict what the value Y_pred will be for any patient given placebo, so that I may write Y_pred=f(X_1,X_2……..X_billion) where f is a function as rich and complicated as you like. Now, if I have observed Y_pred, why should I care what any of these unobserved factors are? Y_pred trumps them all and it is only one factor not a billion. Furthermore, I can measure it directly. All that I require is a probabilistic mode of how it should vary between and within groups. Given randomisation, I have such a model and my analysis relates the way that Y_pred varies within groups to the way that it varies between. Why should I start worrying about less relevant things we haven’t measured. if I should, then don’t just give me words. Don’t just wave your arms and refer vaguely to billions of unmeasured things. Give me a calculation or a simulation that shows it’s a problem. I think, that when you start this exercise, you soon realise that it’s harder than you think.
So I reject any strong claim that RCTs are problematic in this way.
However, I accept the limitations as raised in the second part but these have to be seen as being very debatable reasons for abandoning the advantage of RCTs. There are two reasons. First, higher order effects are plausibly less important than main effects. Strong interactions between subjects and treatments are unlikely if the subject effect is small and the treatment effect is small. But large subject effects are a good argument for paying particular attention to within-study validity, a point at which RCTs excel. Second, large treatment effects are a good argument for questioning whether further study is really in the interests of patients. This brings me on to the second of the arguments. Resources are finite and for me, the question is always, ‘given that the treatment appeared to work in the trials that we ran, and that other treatments are waiting to be studied, does studying this treatment in yet another subgroup make the cut?’. In deciding this, I fully admit that the sort of discussion to be found in the second part of Deaton and Cartwright will be extremely useful.However, I also think that the answer will often be ‘no’.
So, I agree with you that it is strange and wrong that a statistical device should be seen as being as strong as its strongest link. However, I see things rather differently. RCTs bring great strength to reinforcing the weakest link of the argument. That’s the whole point. I fully agree, that this does not make the argument as strong as this link is made. However, it does make it stronger than those arguments that ignore this weakness.
Reference
1. Yates F, Cochran WG. The analysis of groups of experiments. Journal of Agricultural Science 1938;28(4):556-80.
Stephen: Can you explain a bit more your point: “They are allowing that the average treatment effect for the millions of patients that might have been included is zero but then sampling varying effects (that is to say the difference the treatment makes), rather than merely values (that is to say the reading for given patients), from this distribution”.
Pingback: Deaton-Cartwright-Senn-Gelman on the limited value of randomization | LARS P. SYLL
Deborah: In my paper Added Values, I discussed the dispute between Fisher and Neyman thus (bear in mind that the context is agriculture)
“In words, Fisher’s null hypothesis can be described as being, ‘all treatments are equal’, whereas
Neyman’s is, ‘on average all treatments are equal’. The first hypothesis necessarily implies
the second but the converse is not true. Neyman developed a model in which on average over
the field, the yields of different treatments could be the same (if the null hypothesis were true)
but they could actually differ on given plots. Although it seems that this is more general than
Fisher’s null hypothesis it is, in fact, not sensible. Anyone who doubts this should imagine
themselves faced with the following task: it is known that Fisher’s null hypothesis is false and
the treatments are not identical; find a field for which Neyman’s hypothesis is true.” (p 3733)
Reference
1. Senn SJ. Added Values: Controversies concerning randomization and additivity in clinical trials. Statistics in Medicine 2004;23(24):3729-53.
Stephen:
I’ve always thought the Neyman model makes more sense than the Fisher model, as it has never made sense to me to think of treatment effects to be exactly the same for all people. See here for why I say this: http://andrewgelman.com/2013/05/24/in-which-i-side-with-neyman-over-fisher/
Yes, I read this at the time and neither agreed nor disagreed. Basically I think that if you go down that route for testing, you need to look at two one-sided dividing hypotheses and control the error of choosing the worse treatment as ‘better’, rather as proposed by Schwartz and Lellouch (1) over 50 years ago. Of course this immediately leads into decision-analytic considerations but in any case, I don’t think any of it leads to the conclusion that randomisation is a bad idea.
However, to allow for treatment-by-unit interaction in estimation is eminently sensible. Even here, however, there are problems with Neyman’s approach and they carry-over into the apparently endless discussion of random effects meta-analysis, a point that I discussed here (2)
http://onlinelibrary.wiley.com/doi/10.1002/sim.2639/abstract , where I claimed that the combination of large variation in effects and small average effects is rarely very credible.
So to pick up your statement ‘as it has never made sense to me to think of treatment effects to be exactly the same for all people’, in my opinion, that’s really not the point at issue. It is rather that it can be dangerous to allow that treatment effects can be large individually and small on average and the extreme combination of this is to allow that the former are large and the latter is zero
To put it another way, allowing for an interaction and having the main effect null is also a marginality violation. .
References
1. Schwartz D, Lellouch J. Explanatory and pragmatic attitudes in therapeutic trials. Journal of chronic diseases 1967;20:637-48.
2. Senn SJ. Trying to be precise about vagueness. Statistics in Medicine 2007;26:1417-30.
Andrew:
To me and I think Stephen, the definition of Neyman Null Model is that the mean is exactly zero or some constant. If by Neyman Model you take the restriction off the mean, I don’t think there is much, if any disagreement?
Keith O’Rourke
Stephen:
First, I’ve invited Deaton and Cartwright to comment or link us to relevant replies, as I always do when people are discussed, but I don’t know we’ll hear from them (Deaton suggested he was too old for blogs).
Second, I’m traveling back to the states tomorrow, so, while I’ll leave the comments open, anyone who hasn’t been approved before is likely to be held in moderation.
Third, I love your remark:
“RCTs bring great strength to reinforcing the weakest link of the argument. That’s the whole point. I fully agree, that this does not make the argument as strong as this link is made. However, it does make it stronger than those arguments that ignore this weakness.”
There’s an important thing to say about the allegation to the charge that an argument is only as strong as its weakest link. That position is its based on the kind of notion philosophers tend to like, of an argument as a kind of tower, where finding a weak spot causes it all to tumble down. It is used by critics of regulatory positions, ignoring the strong evidence that’s not threatened by the piece they chip away at. That gambit works for a tower or “linked” argument, but not for what’s called a “convergent’ argument wherein many distinct strands work together to build a strong argument from coincidence based on distinct pieces, & self-correcting checks
Anyway, I’m disappointed that you think the issue is analogous to the Neyman-Fisher issue on that experimental design matter, in that Neyman never questioned randomization..
Deborah, you misunderstand me. I agree that Neyman, never disputed the value of randomisation and, was subsequently careful, (and, in view of their dispute, generous) to ascribe the idea to Fisher. I was simply pointing out that F & N have two different null hypotheses and that Neyman’s does not (to me) make any sense.
We have precise control of the allocation algorithm (who gets what if they are in the trial) and virtually none over the presenting process (who gets into the trial) and the null hypothesis that we test is that the effect is zero in the sample not in some fictional population.
(Emphasis mine.) I don’t understand this statement. Isn’t the null hypothesis a statement about a population? And isn’t an effect trivially either zero or non-zero in a particular sample? If you’re only concerned with the sample at hand, what is the point of any kind of statistical test? What are you drawing inferences about?
Noah: Concern is with the causal process that produced aspects of the sample.
It seems like this is related to Kyburg’s concerns over the “reference class” in an inquiry. What is the reference class in a RCT?
The population is the population of all random allocations. For the smallest of the sample sizes that Deaton and Cartwright consider there are more than one hundred trillion such allocations.
It’s only ever the first step. Step 1: ‘prove’ that the treatment worked in the patients actually studied.
These are the questions I suggested in my paper ‘Added Values’ (1) were relevant to thinking about data from clinical trials
Q1. Was there an effect of treatment in this trial?
Q2. What was the average effect of treatment in this trial?
Q3. Was the treatment effect identical for all patients in the trial?
Q4. What was the effect of treatment for different subgroups of patients?
Q5. What will be the effect of treatment when used more generally (outside of the trial)?
For a practical application of this philosophy see Araujo, Julious and Senn (2)
References
1 Senn SJ. Added Values: Controversies concerning randomization and additivity in clinical trials. Statistics in Medicine 2004;23(24):3729-53.
2. Araujo A, Julious S, Senn SJ. Understanding variation in sets of n-of-1 trials. PloS one 2016;11(12):e0167167.
Thank you for the responses, and thank you for the paper references. I have some follow up comments and questions.
With respect to Mayo’s response: My understanding is that it’s the the design of and measurement(s) used in an RCT that enable us to identify causal processes, not the statistical tests used to analyze the data.
With respect to Stephen Senn’s response: Qs 1-4 are sample-specific, right? These can be answered by estimating appropriate statistics (means, SDs) in the sample, and so don’t require statistical tests, as far as I can tell. By way of contrast, Q5 seems to be explicitly about drawing inferences about some population of interest, so it seems inconsistent with the text I quoted and emphasized from the post.
Yes, questions 1-4 are sample specific but no it is not true to say that these do not require statistical tests. The reasoning is like this.
1) A convenience sample is drawn. Its probabilistic relationship to any population of patients is not known. To make inferences about such a population requires the sort of reasoning that forms the second part of Deaton and Cartwright’s two part paper and I specifically excluded that from my discussion. It is the matter of Q5 but I simply listed all the questions covered in my Added Values paper. Q5 is not the subject of discussion here. You will, however, find some comments on it in that paper..
2) The group of patients so sampled are allocated at random to either treatment (T) or control. (C) If we could under identical conditions treat every patient with T and C, then what you say about using the observed difference and not needing statistical tests would be true.
3) However we have allocated the patients at random. This means that the 25 T and 25 C split that we chose is one of 50!(25!25!)=1.264*10^14 possible such splits. We consider the relationship of the actual allocation we had to the population of all the allocations we could have made, since the average of all these populations would give us the average causal effect in the 50 patients, since over all such allocations every patient would be treated half the time with T and half the time with C.
4) Thus we do use inferential statistics to answer Q1 and, with more difficulty, we can also answer Q2. Q3 and Q4 pose further difficulties and depend very much on the design chosen
The whole theory of randomisation and permutation tests is related to this. The book I cited perviously is a good reference. For a simple discussion related to clinical trials see
1. Ludbrook J, Dudley H. Issues in Biomedical Statistics – Statistical-Inference. Aust N Z J Surg 1994;64(9):630-36.
2. Ludbrook J, Dudley H. Why permutation tests are superior to t and F tests in biomedical research. American Statistician 1998;52(2):127-32.
There was a typo. The formula should have been written 50!/(25!25!)=1.264*10^14
This discussion on how to apply the results of RCTs to clinical practice is relevant to the problem of over diagnosis which is a crisis in medicine of a similar magnitude as the replication crisis in science generally. There is a recent example connected to the Sprint study on hypertension.
The Sprint RCT recruited patients with systolic BPs between 130 and 180 and found a ‘statistically significant’ reduction in adverse outcomes. This prompted a conclusion that everyone with a systolic BP above 130 should be treated. This gave rise to controversy unsurprisingly.
There was no attempt to estimate the absolute proportion benefitting at different levels of initial BP. It would probably be minimal at 130 but substantial at 180. I teach this in the Oxford Handbook of Clinical Diagnosis but it seems to be lost on those who prepare ‘evidence-based’ guidelines and who criticise pharmaceutical organisations for promoting over diagnosis.
There is an awareness of relative and absolute risk reduction for risk scores based on multiple risk factors. However these concepts are not applied routinely to RCTs based on single entry criteria. There is also a need for confidence or credibility intervals on these absolute proportions benefitting as opposed to P values based on a null hypothesis.
As far as application to individual patients is concerned, these findings have to be combined with the probability of improved overall well being also bearing in mind adverse effects etc. Decision analysis has been proposed for doing this of course but the usual approach is discussion with or without ‘shared decision aids’.
The latest cry of methodological terrorism:
https://www.bostonglobe.com/ideas/2018/01/21/for-better-science-call-off-revolutionaries/8FFEmBAPCDW3IWYJwKF31L/story.html
Not related to this post, but I thought I’d park the link here.
Is this the same Angus Deaton who got a Nobel prize last year? If so, I’m surprised he misunderstood randomisation. Brave of you to take him on!
Pingback: The RCT controversy | Real-World Economics Review Blog